
A funkcionális genomika irodalmát, a legújabb cikkeket olvasva egyre többször találkozunk a referencia genom fogalmával. Ilyen eset például, amikor ChIP-SEQ eredményeket vagy egyszerűen transzkripciós faktorok kötőhelyeit közlik valami ilyen formában:
hg18:chr3:1263536:1263545:r.
Ez azt jelenti, hogy a humán referencia genom hg18-as összeépítése, összerakása (build) a hármas kromószómán a kezdő és a végpozíció és hogy a reverz (Crick) szálon van. Ez tehát egy koordináta rendszer, amivel ki lehet jelölni egy pontot, vagy egy szakaszt, azaz e koordináták alapján le lehet tölteni (ki lehet vágni) az adott DNS darabot.
A fentiekből az is kiderül, hogy a következő fontos paraméterek vannak:
1. Hányas számú genomösszerakás
2. Melyik kromoszómán
3. Melyik pozíció
4. Melyik szál
Mi az a genomösszerakás (build)?
Ismert, hogy a 2001-ben amikor közölték a két vázlatos (draft) humán genomot, mindkét esetben a szekvenciát több (férfi) mintából határozták meg (bár már akkor is suttogták, hogy a Celera-s mintában Craig Venter DNS-e is benne volt). Tehát nem egy egyén DNS-ét határozták meg, hanem többét, amit ráadásul a testi kromoszómák esetében szorozni is kell kettővel, mivel nyilvánvaló, hogy édesapánktól és édesanyánktól minden egyes testi kromoszómából két különbözőt kapunk (sőt, egyre inkább úgy tűnik, hogy nagyon is különbözőket). Gondolom az is nyilvánvaló, hogy hiába a humán genom az egyik legjobban tanulmányozott, azért még nem tartunk ott, hogy lenne a 24-féle kromoszómának megfelelő 24 darab egybefüggő DNS szekvenciánk. Ehelyett, mint az a Genom Referencia Konzorcium honlapján látható;
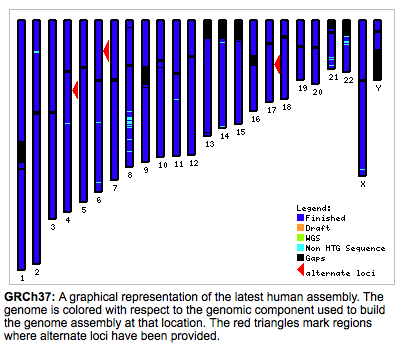
valójában az összes kromoszómán vannak még rések, problémás részek. Mi okozza ezeket? Leginkább a centromérák környéke, vagyis olyan DNS szakaszok, amelyeket nehéz megszekvenálni (pl hosszú homopolimerek, ismétlődő szekvenciák). Emiatt folyamatosan javítják az összerakott szekvenciát (a hg18-ból ki lehet találni, hogy megélt már 18 összerakást), ami így nem egy valódi kromoszómaszekvencia, hanem egy úgynevezett pszeudokromoszóma, ahol a réseket (és a kromoszómavégeket) N-ekkel töltik ki.
Miért fontos tudni, hogy a hg18-ról van szó?
Azért, mert tegyük fel, hogy 2009 elején csinálnak egy egér ChIP-SEQ kísérletet, ahol transzkripciós faktorok által fedett DNS darabokat szekvenálnak, majd az eredményeket ráillesztik az akkor elérhető mm8-as egér genomszekvenciára. A közölt koordináták alapján ellenőrizni akarjuk ma valamelyik fedett régió szekvenciáját, ezért a koordináták alapján letöltjük a szekvenciát a mostani mm9-es genomszekvenciából. Könnyen előfordulhat, hogy egy teljesen más szekvenciát fogunk letölteni, mint amivel a szerzők dolgoztak!
Miért?
Azért mert ha pl. az adott kromoszóma elején találtak egy nem odaillő szakaszt, és azt kivették az mm9-esben, akkor a koordináták annyival elcsúsznak! Tehát nagyon fontos, hogy mindig nézzük meg, hogy melyik genomösszerakásból származnak a koordináták, és abból szedjük ki a szekvenciát! Ez a gyakorlati bioinformatikai munkában nagyon sok gondot okoz, ezért én például azt preferálom, hogy nem a pozíciót, hanem a szekvenciát tárolom (SNP-nél vagy transzkripciós starthelynél pedig az adott pont körüli 50-50 bp szekvenciát), és ha pozíció kell (mert például a pozíciók alapján keresek átfedést egy másik eredménnyel), akkor visszaillesztem az éppen aktuális genomra 100% azonossági küszöbbel).
Ez szerintem egyre nagyobb problémát jelent, hiszen most már nagyon sok funkcionális genomikai eredmény érhető el a régebbi genomösszerakásokra, ráadásul a „többi” faj genomszekvenciáit is elkezdik újraszekvenálni, tehát sok genomot fognak újra összerakni.
Összefoglalva:
A referencia genom a haploid genom (embernél a 24-féle kromoszóma + a mitokondrium DNS) pszeudoszekvenciáit tartalmazó konszenzus szekvenciák összessége, ami önmagában ilyen formában nem létezik és nem is létezett. A célja az, hogy a többi szekvenciát ehhez viszonyítsuk. A referencia genomszekvenciákat folyamatosan újraépítik, ami azt jelentheti, hogy a szekvencia koordináták elcsúszhatnak, ezért vigyázni kell, hogy ha nem szekvencia, hanem koordináta alapon keresünk például két szekvencia között átfedést, akkor ugyanazt az összerakott genomot használjuk (pl. hg18 vs hg18 vagy mm8 vs mm8).
A referencia genomszekvenciákat a nagy genomikai portálokról lehet (érdemes) letölteni. Javaslom az ensembl, az NCBI vagy a személyes favoritom, az UCSC webhelyét.


 eszembe jutott, hogy megírtam félig egy posztot egy hónapja (időközben több helyen is, például az
eszembe jutott, hogy megírtam félig egy posztot egy hónapja (időközben több helyen is, például az  Következő reggel mikor a szüleimnél ismét internet közelbe kerültem és elolvastam az elektronikus leveleimet, láttam meg hogy a JGI-ben
Következő reggel mikor a szüleimnél ismét internet közelbe kerültem és elolvastam az elektronikus leveleimet, láttam meg hogy a JGI-ben  amely szinte minden évben elfagy (igaz tavaly például a jég verte el), szóval biztos tudna a genomika segíteni egy fagyállóbb fajta kinemesítésében például.
amely szinte minden évben elfagy (igaz tavaly például a jég verte el), szóval biztos tudna a genomika segíteni egy fagyállóbb fajta kinemesítésében például. (gyermekkoromban nyaranta mindig a zalakarosi szőlőhegyen nyaraltam, és emlékszem ott is volt egy gyönyörű öreg gesztenyefa, bár a közelébe se ért ennek). Sikerült is megtalálni, és ráadásul olyan szerencsénk volt, hogy terület egyik tulajdonosa éppen ott volt, így megtudhattuk tőle többek közt, hogy legalább ötszáz éves a fa, és jóval ellenállóbb a betegségekkel szemben, mint a mellette álló fiatal testvére. Ezen egyből el is gondolkodtam, hogy érdemes lenne itt is összehasonlítani egy mai fiatal, és ennek a matuzsálemnek a genomját, és átgondolni, hogy mi a jelentősége evolúciós szempontból, hogy egy fa ötszáz évig hullajtja magjait.
(gyermekkoromban nyaranta mindig a zalakarosi szőlőhegyen nyaraltam, és emlékszem ott is volt egy gyönyörű öreg gesztenyefa, bár a közelébe se ért ennek). Sikerült is megtalálni, és ráadásul olyan szerencsénk volt, hogy terület egyik tulajdonosa éppen ott volt, így megtudhattuk tőle többek közt, hogy legalább ötszáz éves a fa, és jóval ellenállóbb a betegségekkel szemben, mint a mellette álló fiatal testvére. Ezen egyből el is gondolkodtam, hogy érdemes lenne itt is összehasonlítani egy mai fiatal, és ennek a matuzsálemnek a genomját, és átgondolni, hogy mi a jelentősége evolúciós szempontból, hogy egy fa ötszáz évig hullajtja magjait.
Utolsó kommentek